抗淀粉样蛋白聚集的铂类抑制剂的设计与合成毕业论文
2020-07-09 20:27:05
摘 要
阿尔茨海默病(Alzheimer's disease, AD)是最常见的神经系统退行性疾病。该疾病具有高致死率。AD的主要特征表现为不可逆转性和进行性的认知功能障碍。引起AD的主要原因是十分复杂的并且包含着诸多的风险因素,主要包括遗传因素、环境因素和生活方式等。尽管AD确切的致病机制还没有被完全解释清楚,但人们通过在生物化学、细胞病理学、分子水平以及遗传学水平方面的研究增加了对该疾病的了解。当前,有诸多的病理假说被提出来,其中主要包括胆碱能假说、金属离子假说、氧化应激假说、β淀粉样蛋白(Aβ)级联假说等。同时也有很多重要的证据表明,Aβ的聚集是AD发生和发展的关键事件和致病因素。各种Aβ聚集体,包括寡聚体,原纤维和纤维等,特别是寡聚体可能会引发一系列的致病级联,从而导致神经元死亡、突触功能障碍和认知障碍等,并最终导致AD的产生。因此在这一方面,Aβ聚集体已经被认为是AD诊断和治疗的生物标志物和药物靶标。近年来研究发现铂(II)金属配合物能够通过铂金属与Aβ中的组氨酸配位作用相结合,从而抑制Aβ聚集。但是由于毒副性以及相对差的血脑屏障通过性限制了其进一步应用。铂(IV)配合物因其惰性特性且需激活后才能显示活性的特点具有无毒和定点激活等优势。在此基础上,本课题拟设计、合成一类新型的铂(IV)配合物,使其既具有Aβ靶向功能、又能够抑制Aβ聚集,同时可以通过自身荧光变化识别并跟踪检测Aβ的聚集程度,从而为AD的诊断和药物治疗研究提供新的思路与方法。
关键字:阿尔茨海默病 Aβ聚集 金属离子 铂类配合物
ABSTRACT
Alzheimer's disease (AD) is the most common neurodegenerative disease. The disease is extremely destructive and can sometimes cause death. The main characteristic of AD is irreversible and progressive cognitive dysfunction. The main cause of AD is very complicated and contains many risk factors, including genetic factors, environmental factors and lifestyle. Although the exact pathogenesis of AD has not yet been fully elucidated, studies on biochemistry, cytopathology, molecular level, and genetics have increased people's understanding of the disease. To date, there are many hypotheses that have been proposed, mainly including the cholinergic hypothesis, metal ion hypothesis, oxidative stress hypothesis, and β-amyloid(Aβ) cascade hypothesis. A large number of important evidences indicate that the aggregation of Aβ is a key event and pathogenic factor in the occurrence and development of AD. Various Aβ aggregates, including oligomers, fibrils, and fibers, especially oligomers, may initiate a series of pathogenic cascades that lead to neuronal death, synaptic dysfunction, and cognitive disorders, and eventually lead to AD. In this regard, Aβ aggregates have been considered as biomarkers and drug targets for the diagnosis and treatment of AD. In recent years, it has been found that platinum (II) complexes can bind to Aβ via the coordination between platinum(II) and histidine residues in Aβ, thereby inhibiting Aβ aggregation. However, due to toxic side effects and the relatively poor blood-brain barrier permeability, further applications of platinum(II) complex are limited. Platinum (IV) complexes have the advantages on non-toxicity and activation at disease site due to their inertness and activated activities. Accordingly, this thesis intends to design and synthesize a new class of platinum (IV) complexes that not only have Aβ targeting function, but also can inhibit Aβ aggregation. They can also identify and track the aggregation of Aβ through the change of their fluorescence simultaneously. This thesis provides new ideas and methods for AD diagnosis and drug treatment research.
KEYWORD:Alzheimer's disease; Aβ aggregation; Metal ions; Platinum complexes
目录
摘要 I
ABSTRACT II
目录 III
第一章 绪论 1
1.1 引言 1
1.2 AD的发病机制 1
1.2.1淀粉样蛋白级联假说 1
1.2.2金属离子假说 2
1.2.3氧化应激假说 2
1.3 β-淀粉样蛋白 3
1.3.1 Aβ的产生 3
1.3.2 Aβ的降解 3
1.4 金属螯合剂 4
1.5 铂类金属配合物对Aβ肽聚集的抑制 7
1.6研究意义 9
第二章 实验部分 10
2.1 主要试剂及仪器 10
2.1.1 化学试剂 10
2.1.2 实验仪器 10
2.2 Pt(BTA)Cl4的制备 11
2.2.1 T-S的合成 11
2.2.1 BTA的合成 12
2.2.3 BTA-Pt的合成 13
2.2.4 Pt(BTA)Cl4 的合成 13
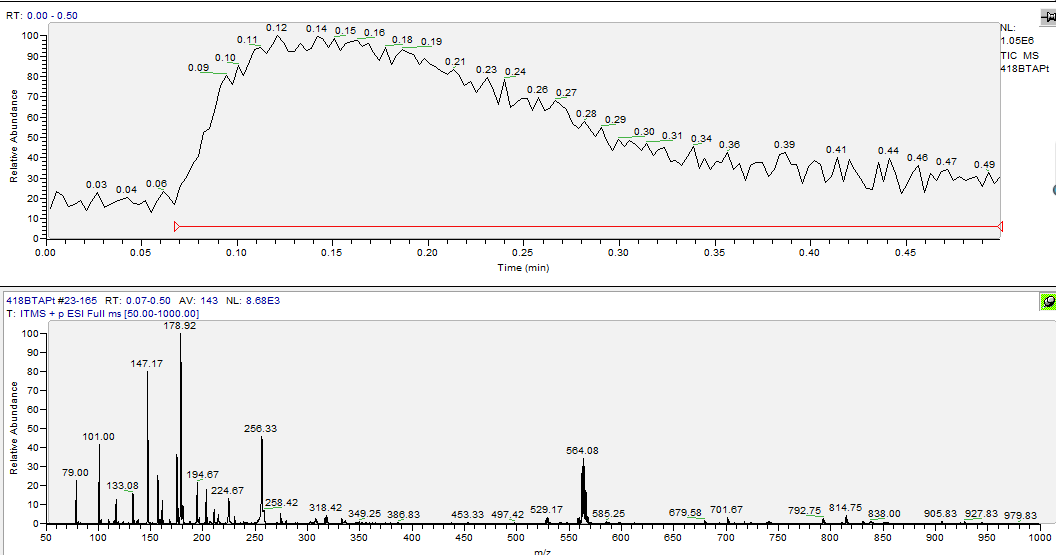
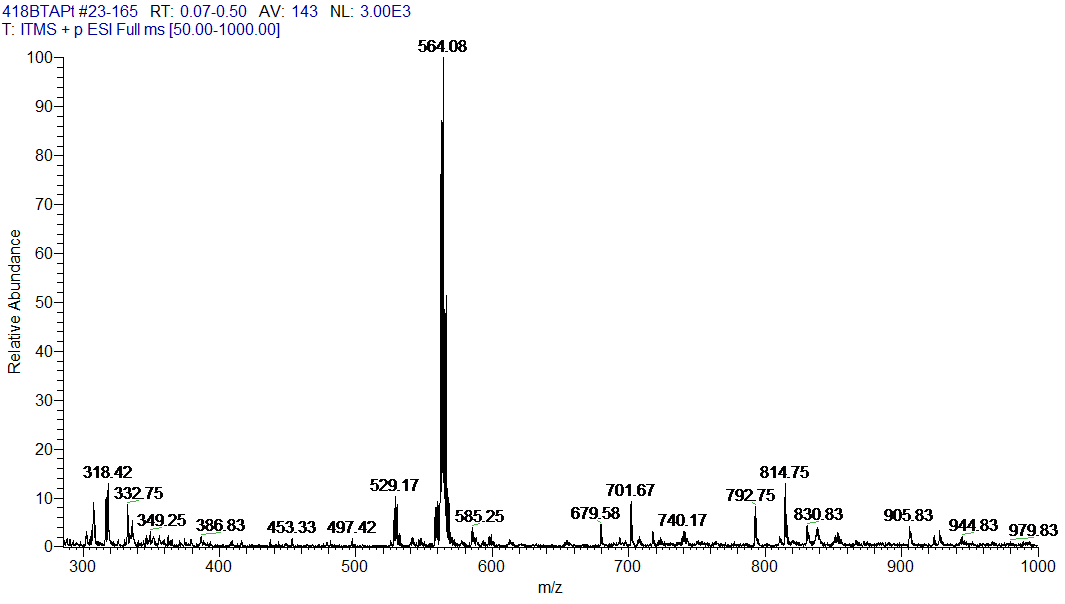
第三章 结果与表征 16
3.1 T-S的表征与分析 16
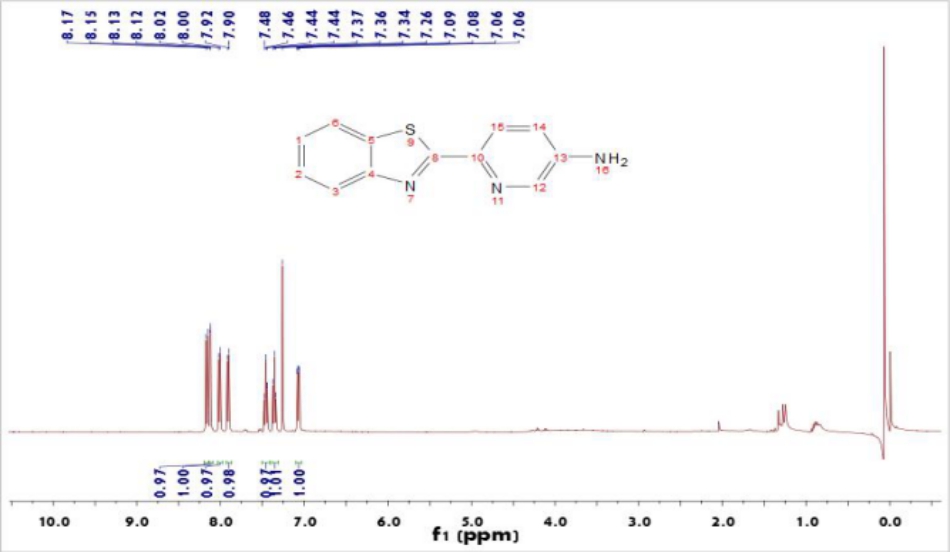
3.2 BTA的表征与分析 17
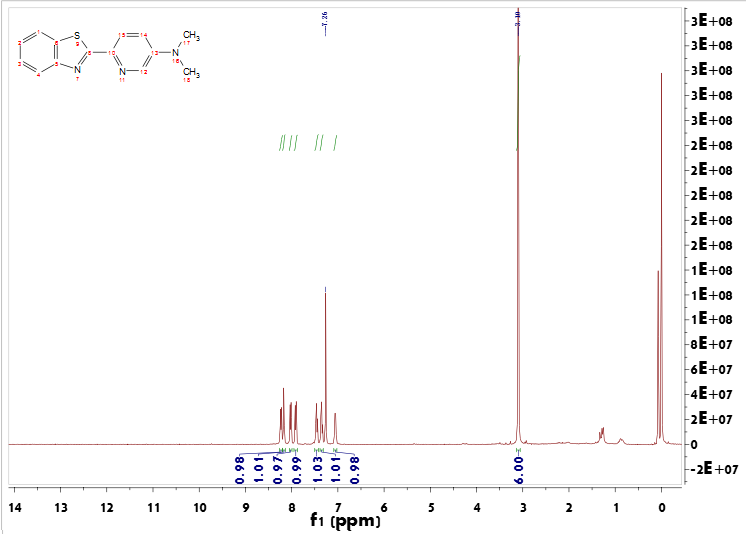
3.3 BTA-Pt的表征与分析 18
3.4 [Pt(BTA)Cl4]的表征与分析 20
3.4.1 [Pt(BTA)Cl4]第一次合成的表征与分析 20
3.4.2 [Pt(BTA)Cl4]第三次合成的表征与分析 22
3.4.3 [Pt(BTA)Cl4]第四次合成的表征与分析 23
第四章 结论与展望 24
4.1结论 24
4.2 展望 24
参考文献 25
致谢 30
第一章 绪论
1.1 引言
阿尔茨海默病(Alzheimer' s diseasea , AD)是痴呆的一种,其发病原因比较复杂,且与人的年龄大小有着密不可分的关系,是一种不可治愈性的疾病。伴随着年纪的增长,其发病的概率也会随之增加。据统计,AD患者发病的分界年龄点一般为65岁,并以此为基础可将其分为两种类型:一种为早发型AD(EOAD),另一种为晚发型AD(LOAD)[1]。早发型AD只占所有病例中极小的一部分,一般约为5%,这种类型的AD一般与遗传因素脱不了关系;而大多数的AD病例为晚发AD,并且是散发性的[2]。在美国,65岁以上的老人中有13%的人患有阿尔茨海默病,但85岁以上的老人中,这一比例则迅速上升到了40%,并且他们的平均的生存期为确诊后的8年[3]。AD患者的主要临床表现为记忆力降低、思维能力和语言功能障碍、感知能力下降、方向感缺失、性格改变以及失去独立性等[4],也会伴随出现幻觉、妄想、甚至焦虑、抑郁等症状,这些都已经严重危害到了老年患者的健康与生活品质。AD典型的病理学特征一般可概括为三个方面:(1)大脑皮质出现由β淀粉样蛋白(Amyloid protein, Aβ) 聚集形成的老年斑(senile plaques, SP);(2)神经纤维缠结(neurofibrillary tangles, NFT);(3)胆碱能神经元缺失[5]。此外,大量与病理学、生化学有关的证据支持Aβ聚集在AD发病机制中起主导作用。因此,已经将消除Aβ的聚集体成为治疗AD的潜在方法。
1.2 AD的发病机制
关于AD在生理病理学上的第一种解释即为胆碱能假说,该种假说是以神经递质中乙酰胆碱水平的降低为基础的,这也就意味着其与中枢神经系统(CNS)的记忆和学习有关[6]。该假说指出,AD主要是由缺乏乙酰胆碱引起的。因此,抑制乙酰胆碱酯酶(AChE)成为了治疗AD的主要方法[7,8]。但伴随着生物无机化学在AD中发展的作用,出现了三种与金属相关的假说,并且引起了人们对AD发病机制的阐述,包括淀粉样蛋白级联假说、金属离子假说和氧化应激假说。[9-11]
1.2.1淀粉样蛋白级联假说
淀粉样蛋白级联假说推测淀粉样蛋白-β肽(Aβ)在脑中的异常聚集和沉积是AD中的起始因子和关键事件,其引发了一系列异常的级联并导致细胞死亡,最终导致AD[12-13]。Aβ(长度为39-43个氨基酸)源自β-分泌酶和γ-分泌酶对淀粉样蛋白前体蛋白(APP)的切割,其包含亲水性N-末端和疏水性C-末端。最丰富的片段是Aβ40和Aβ42。Aβ产生和清除之间的不平衡导致其聚集成为寡聚体、原生质体、膜和最终老年斑,这是AD的主要组织病理学标志之一[14]。 Aβ聚集体,特别是寡聚体,具有神经毒性,是动物的病理触发因素。C-末端和中央疏水残基Leu17-Ala21,也称为自我识别残基,在通过疏水相互作用和氢键形成Aβ聚集体时很重要。然而,聚集通常受温度、pH、金属离子等影响。由于N-末端含有能够配位过渡金属离子(例如Cu(II)、Fe(III)和Zn(II))的金属结合位点,同时因为其高的亲和力,从而使得金属离子极大地影响了Aβ的总体聚集行为[9,15]。这些位点的金属结合可以加速Aβ聚集,并产生更多的非晶聚集体,包括神经毒性寡聚体。金属离子与Aβ的详细结构相互作用已经在几篇综述中被描述[7,19,20],尽管一些争论仍然存在,例如,大脑中Aβ沉积数量与认知障碍程度之间的相关性较差,但淀粉样蛋白级联假说依然在对研究AD的发病机制方面产生了深远的影响。根据这一假设,Aβ聚集体被认为分别作为AD的诊断和治疗的生物标志物和药物靶标[16]。在这方面,已经开展了大量努力来制定适当的治疗策略,目的是减少Aβ产生,抑制Aβ的聚集,加速抗炎或增强抗体清除率[17,18]。
1.2.2金属离子假说
金属离子假说侧重于另一主要病理学特征,金属稳态受损,尤其是Zn、Cu和Fe,该假说认为金属稳态失衡可能有利AD进展[9,19]。锌,铜和铁是维持大脑正常神经功能的重要元素。然而,在AD脑组织中的几个皮质下区域观察到了异常水平。正如在淀粉样蛋白级联假说中所提到的,金属离子与Aβ的相互作用对稳态失衡起着重要的作用,它会导致淀粉样斑块中Zn(约1 mM)、Cu(约0.4 mM)和Fe(约1mM)的异常高浓度[20]。此外,其他与AD相关的蛋白,包括APP、tau和早老素,也可能与AD中金属水平和体内平衡的变化有关。因此,这种假设有利于治疗性干预,可能会抑制AD大脑中的金属稳态。
1.2.3氧化应激假说
在氧化应激假说中,广泛的氧化应激是AD大脑的一个特征,被认为是AD的致病因素。氧化应激反映了一种不平衡,这种不平衡性主要体现在活性氧(ROS)的产生与抗氧化防御系统之间,他们损害了线粒体的功能,导致了神经元的凋亡,并进而引发了AD的认知障碍[21]。ROS的产生与氧化还原活性金属离子,特别是Cu(II)和Fe(III)金属离子有着强烈的关系。 像Cu(I)/ Cu(II)和Fe(II)/ Fe(III)之类的氧化还原对之间的氧化还原循环可促进分子氧的活化,其主要由Aβ介导。Cu(II)和Fe(III)Aβ配合物能够在氧气存在下通过还原剂还原而催化还原生成每个氧化物(H2O2)的氢。 通过与H2O2的芬顿型反应,金属蛋白质以还原形式催化高毒性羟基自由基的生成[22]。 此外,谷胱甘肽是一种有效去除ROS的纤维素抗氧化剂,铜稳态失衡可能通过降低其水平来增强ROS的细胞毒性[23]。 因此,该假设认为抗氧化剂应该对AD疗法有一些有益的效果。
1.3 β-淀粉样蛋白
1.3.1 Aβ的产生
Aβ是通过淀粉样蛋白前体蛋白(APP)的连续蛋白水解切割而产生的[24,25]。Aβ斑块伴随有神经毒性和氧化应激,其会在人脑中产生具有脂质过氧化的自由基。淀粉样前体蛋白(APP)通常会被两条途径所切割:淀粉样蛋白形成途径和非淀粉样蛋白形成途径[26]。在前一条淀粉样蛋白的形成途径中,β-和γ-分泌酶的切割能够引发神经毒性Aβ的产生,即APP先被β-分泌酶(也可以称为β-位点APP切割酶(BACE)切割并产生跨膜C端片段β(CTFβ)[27]。然后用γ-分泌酶裂解CTFβ产生Aβ,其中γ分泌酶由四种膜蛋白质组成,即早老蛋白1(PS1)、早老蛋白2(PS2)、呆蛋白和前咽缺损[28]。在后一种非淀粉样蛋白途径中,APP被α-分泌酶裂解,释放出可溶性N-末端片段(sAPPα)和剩余的C-末端片段(αCTF)。γ-分泌酶随后裂解αCTF,导致非淀粉样多肽P3肽(Aβ[17-40]或Aβ[17-42])和APP细胞内结构域的形成[29]。这两种途径都可以在正常生物体中发生。因此,尽管有些含糊不清,但释放的Aβ被认为在健康的大脑中具有一定的功能。然而,一旦其产量和清除率之间的平衡被一些病理因素破坏,如低p H缺氧、高胆固醇血症和锌(II)缺乏[30],Aβ肽就容易聚集。
1.3.2 Aβ的降解
在人脑中,Aβ的降解与清除主要通过胰岛素降解酶(IDE)和中性溶酶(NEP)来完成。在大脑正常情况下,Aβ肽能够被大脑中特有的巨噬细胞与小胶质细胞所摄取,在这个时候就需要借助中性溶酶(NEP)和胰岛素降解酶(IDE)来维持Aβ的稳态[31]。NEP是Aβ降解的限速酶,在NEP基因缺失或者其活性受到抑制这两种情况下,均会导致Aβ聚集的增多,但转染NEP基因能够显著地减少AD小鼠脑内Aβ斑块的聚集[32]。大部分IDE存在于胞质溶胶中,而在线粒体、过氧化物酶体和质膜中存在较少量,IDE的一小部分会被运输到细胞外空间以与IDE的已知底物如胰岛素和Aβ相互作用。在IDE转染的细胞系中细胞外分泌的Aβ也有所减少。最近报道表明,在这些降解过程中,ATP结合盒转运蛋白A1(ABCA1)和载脂蛋白E(ApoE)的存在是不可缺少的[33]。ABCA1是一种作用显著的调节剂,它可以调节全身的胆固醇代谢,并可以将外周组织中多余的胆固醇移至肝脏中,另外其还可增加血浆中的高密度脂蛋白(HDL)水平[34]。通过促进胆固醇流出,ABCA1也介导脂化ApoE和ApoE-HDL颗粒的形成。ApoE是一种载体蛋白,其存在于中枢神经系统(CNS)中,并且可以由中枢神经系统分泌出星形胶质细胞以及小胶质细胞。虽然ApoE的异构体之一ApoE4被认为会增加迟发型AD的发病风险,但ApoE能够促进Aβ的运输,并能够通过ABCA1的帮助将其从脑中清除[35]。缺乏ABCA1可能会降低脂化载脂蛋白E13,并增加可溶性和不溶性Aβ水平,加重AD小鼠模型中Aβ沉积。
1.4 金属螯合剂
金属离子高度参与AD的致病途径,特别是金属诱导的Aβ聚集和氧化应激与金属异位症可能导致神经元细胞死亡,突触功能障碍和认知障碍[36]。事实上,基于这些情况,涉及金属的AD治疗策略已经受到了人们的广泛关注。同时也已经开发了一些能够靶向金属诱导的Aβ聚集体(金属-Aβ)的金属螯合剂[37,38],通过在体外和体内螯合Aβ-结合的金属离子来调节金属动态平衡,Aβ的聚集以及其神经毒性,迄今为止,已经开发了大量的金属螯合剂作为AD中金属螯合治疗的治疗剂[39]例如去铁敏(DFO)、Clioquinol(CQ)和8-羟基喹啉衍生物(PBT2)等。尽管CQ和PBT2在临床研究中失败了,但这种研究通过改进结构修饰的性质而推动了AD治疗用螯合剂的持续发展。事实上,基于多目标定向药物设计策略的多功能金属螯合剂[40]被认为是用于进一步开发AD治疗药物的有前途的主导剂。 因此,通过将诊断成像功能添加到治疗性金属螯合剂支架中来设计基于金属螯合剂的AD诊断剂是合理的。
众所周知,一些淀粉样蛋白结合染料,包括刚果红(Congo red),硫代黄素T(ThT)和姜黄素,都能够通过敏感的荧光反应或成像识别Aβ物种,这可以赋予基于金属螯合剂的诊断药物具有Aβ靶向和荧光成像特性。值得注意的是,近红外荧光成像作为一种有吸引力的诊断工具对于AD的体内应用特别有价值,这主要归因于其无创性操作、可接受的穿透深度以及对生物膜斑自动荧光的最小干扰[41]。因此,与金属螯合支架和Aβ结合染料结合的荧光螯合剂被认为是有希望用于AD治疗的候选物。
相关图片展示:

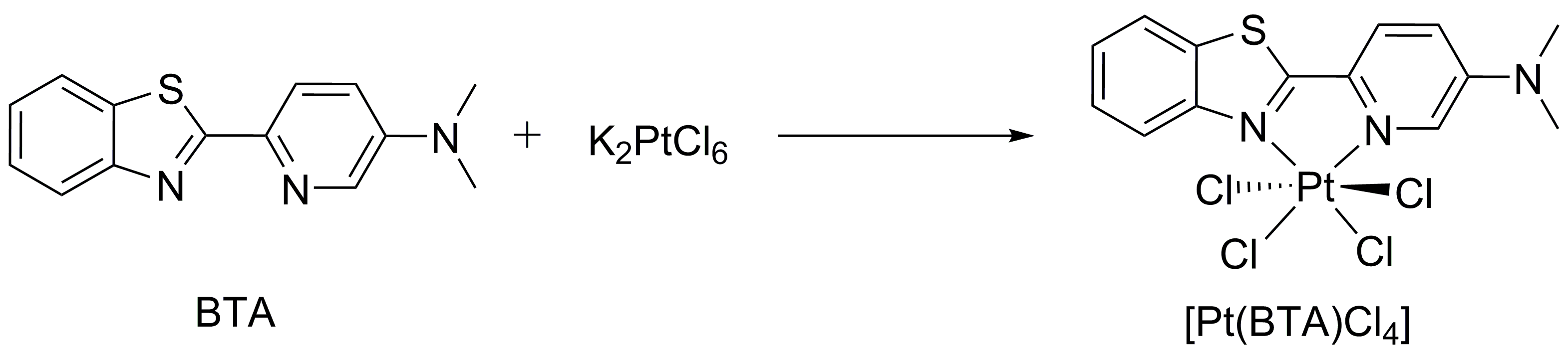
课题毕业论文、开题报告、任务书、外文翻译、程序设计、图纸设计等资料可联系客服协助查找。


